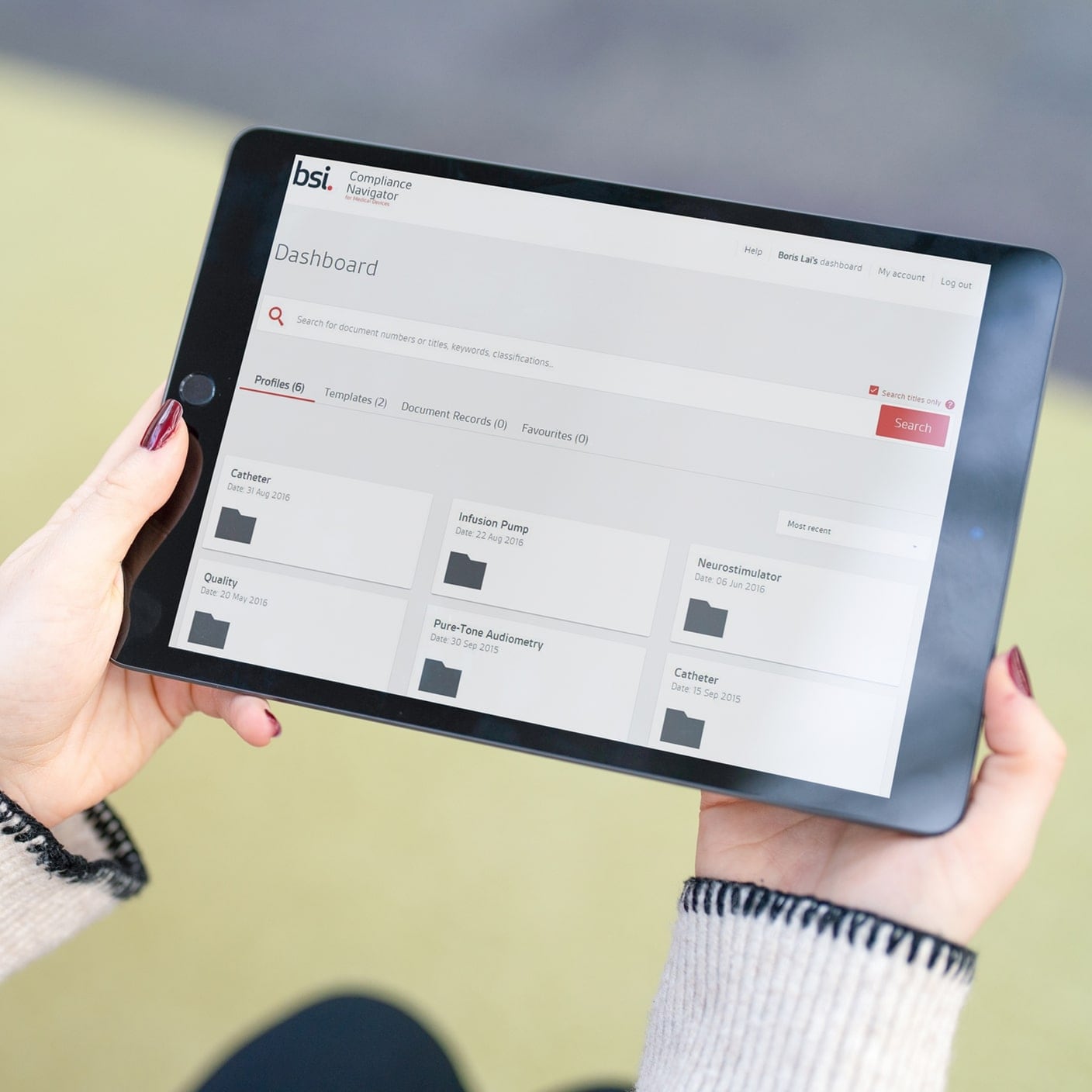
BSI est composé de 5 000 personnes soutenues par 12 000 experts de l’industrie dans plus de 193 pays. Nos services réglementaires, combinés à notre expérience de premier plan, offrent des voies efficaces pour la mise sur le marché de votre dispositif.
Retour au menu
Sélectionner un pays/une région
- Australia - English
- Austria - Deutsch
- Belgium - Nederlands
- Brazil - Português
- Canada - English
- Canada - Français
- Chile - Español
- China - 简体中文
- Colombia - Español
- Costa Rica - Español
- Czech Republic - English
- Germany - Deutsch
- Hong Kong (SAR China) - English
- India - English
- Indonesia - English
- Ireland - English
- Israel - English
- Italy - Italiano
- Japan - 日本語
- Korea - 한국어
- Malaysia - English
- MEA - عربي
- MEA - English
- Mexico - Español
- Mongolia - English
- Netherlands - English
- Netherlands - Nederlands
- New Zealand - English
- Peru - Español
- Philippines - English
- Poland - Polski
- Singapore - English
- South Africa - English
- Spain - Español
- Sweden - English
- Switzerland - Deutsch
- Taiwan - 繁體中文
- Thailand - ไทย
- Turkey - Türkçe
- United Kingdom - English
- United States - English
- Vietnam - English
- Vietnam - Tiếng Việt
Suggested region and language based on your location
Your current region and language